
Pon mucho color en tus gráficos
Irene Soler PhD candidate at CIPF Carla Perpiñá PhD candidate at CIPF La visualización de datos biológicos es la transformación de estos en figuras, con el objetivo de simplificar los […]

Irene Soler PhD candidate at CIPF Carla Perpiñá PhD candidate at CIPF La visualización de datos biológicos es la transformación de estos en figuras, con el objetivo de simplificar los […]
Ana Díaz-de Usera PhD candidate at Universidad de La Laguna Desde que en los años 20 P.A. Levene definiera el ADN como una molécula constituida por cuatro bases nitrogenadas, un

Zulema Rodríguez Hernández PhD student at National Center for Epidemiology Importancia de Cytoscape El avance en la última década de las ómicas ha producido un crecimiento exponencial de datos biológicos.
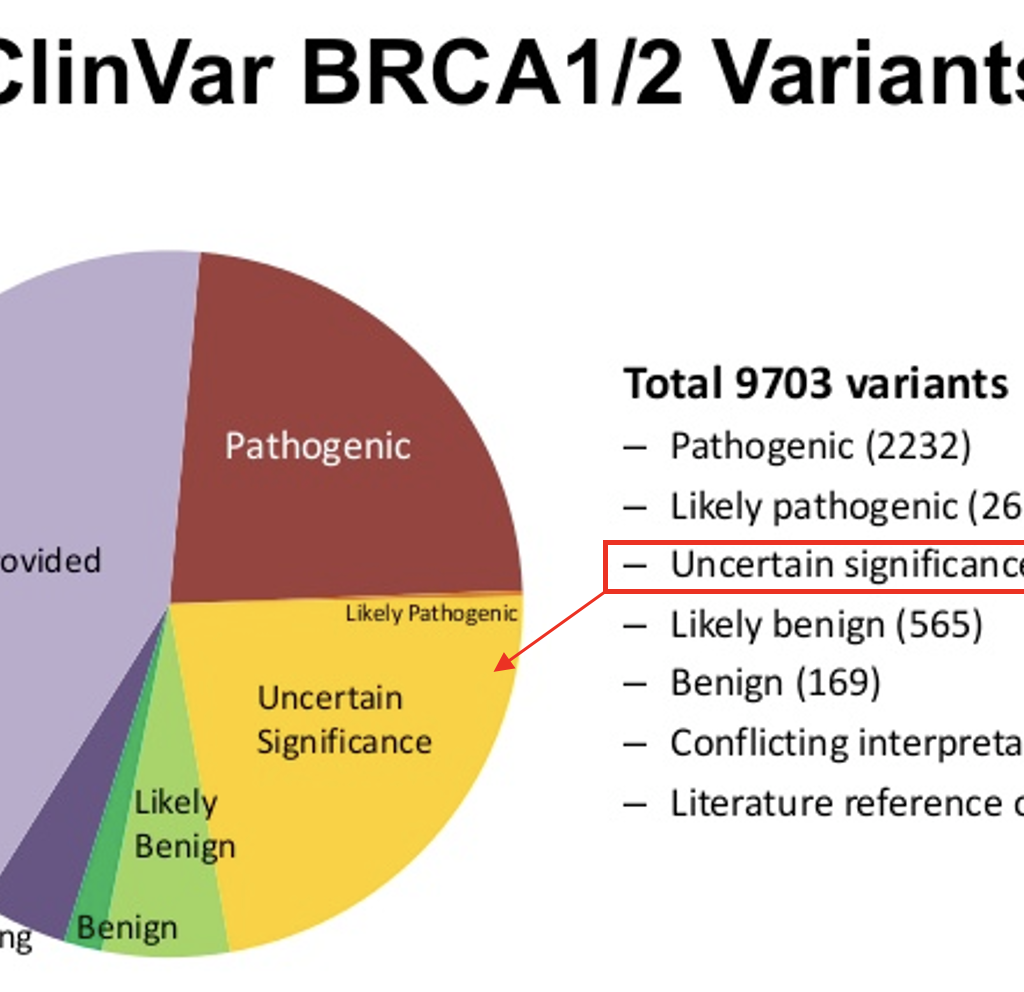
Alejandro Mendoza-Alvarez PhD candidate at Universidad de La Laguna La aparición de los secuenciadores masivos que permiten leer en paralelo de millones a miles de millones de secuencias o fragmentos
Tamara Hernández Beeftink PhD candidate at Universidad de La Laguna Luis A. Rubio Rodríguez PhD candidate at Universidad de La Laguna El pasado mes de Octubre de 2021 (18 y
Inés Rivero Ph.D. Student, Centro Nacional de Investigaciones Cardiovasculares (CNIC) Muchos estudiantes de biociencias habrán escuchado alguna vez la frase “Deberías plantearte una carrera en bioinformática, eso te abrirá muchas

Eva Tosco-Herrera PhD candidate at Hospital La Candelaria When people ask me what I wanted to be as a grown-up when I was little, I honestly do not know what
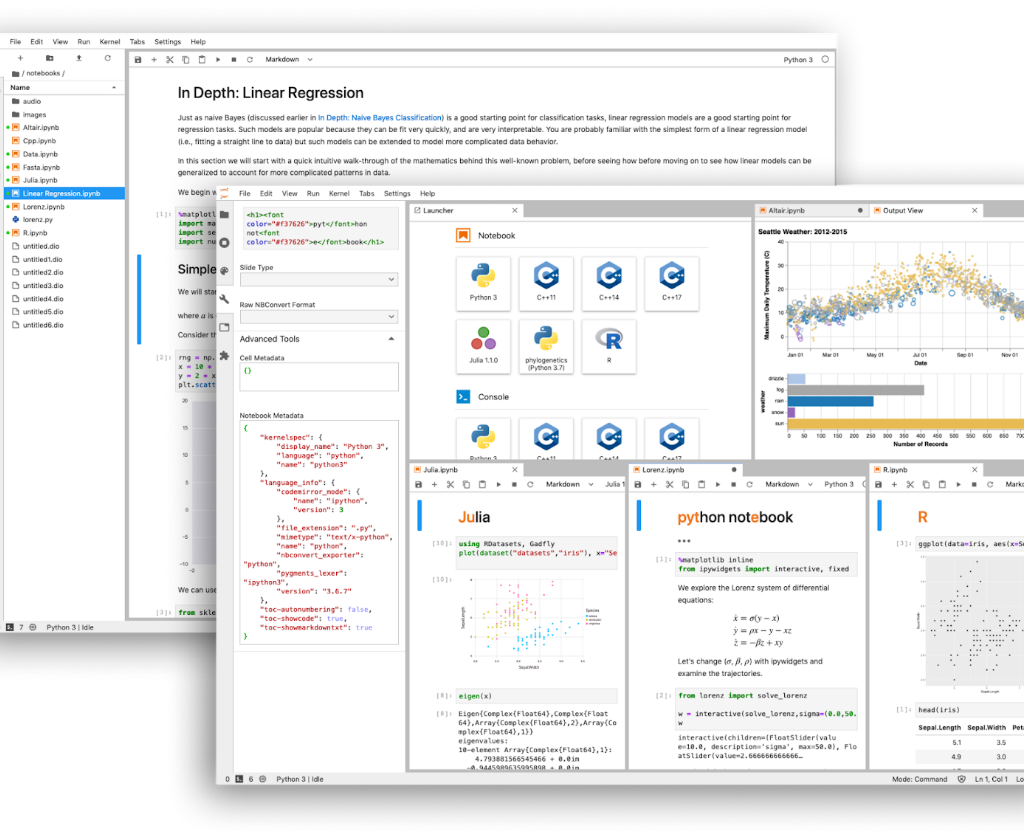
Héctor Rodríguez PhD candidate at Hospital La Candelaria Eva Suárez PhD candidate at Hospital La Candelaria A menudo, para el análisis bioinformático trabajamos directamente con consola, editores de texto para

Sarai Varona Bioinformatician at ISCIII Me llamo Sarai, tengo 26 años, estudié la carrera de Bioquímica y Biología molecular y estudié dos másters, uno en Bioinformática y otro en Biología
VIII Bioinformatics Student Symposium Organizers Online 18-19 October 2021 The VIII Bioinformatics Student Symposium is organized by RSG-Spain, an association of junior scientists in Bioinformatics based in Spain and satellite group