

La aparición de los secuenciadores masivos que permiten leer en paralelo de millones a miles de millones de secuencias o fragmentos del ADN (reads), lo que ha revolucionado el campo de la biología y, especialmente, el de la genética, la cual ha pasado de un ámbito tradicional como es el laboratorio a uno computacional, con la aplicación ineludible de la bioinformática. El desarrollo de estas técnicas de secuenciación masiva de ADN (NGS) ha facilitado enormemente la detección de la variación genética y su estudio en el contexto clínico. Sin embargo, la secuenciación masiva también genera una gran cantidad de información, complicando la detección rápida y sencilla de las variantes genéticas responsables de enfermedades.
Para asistir en el estudio e interpretación clínica de cada una de las variantes genéticas detectadas en la secuenciación masiva, el Colegio Americano de Medicina Genética y Genómica (ACMG) ha establecido un sistema de clasificación para facilitar su asociación clínica. Este sistema se basa en la agrupación de las variantes genéticas en base a 5 clases de patogenicidad: benigna (B), probablemente benigna (LB), variante de significado incierto (VUS), probablemente patogénica (LP) o patogénica (P). Uno de los principales desafíos a nivel mundial sigue siendo resolver el potencial patogénico de una alta proporción de variantes de significado incierto (VUS).

Desgraciadamente, en la actualidad, la gran frecuencia de variantes que no se decantan por ninguno de los dos lados de la balanza llegan a alcanzar cifras tan alarmantes como un 80% de variantes clasificadas como VUS por estudio de secuenciación masiva, tal y como ocurre en el caso de los genes BRCA1 y BRCA2, conocidos ampliamente por su asociación con el cáncer de mama y ovarios, entre otros. Llama la atención que, pese a ser dos de los genes más estudiados y secuenciados, la proporción de variantes de significado incierto (VUS) es sorprendentemente alta.
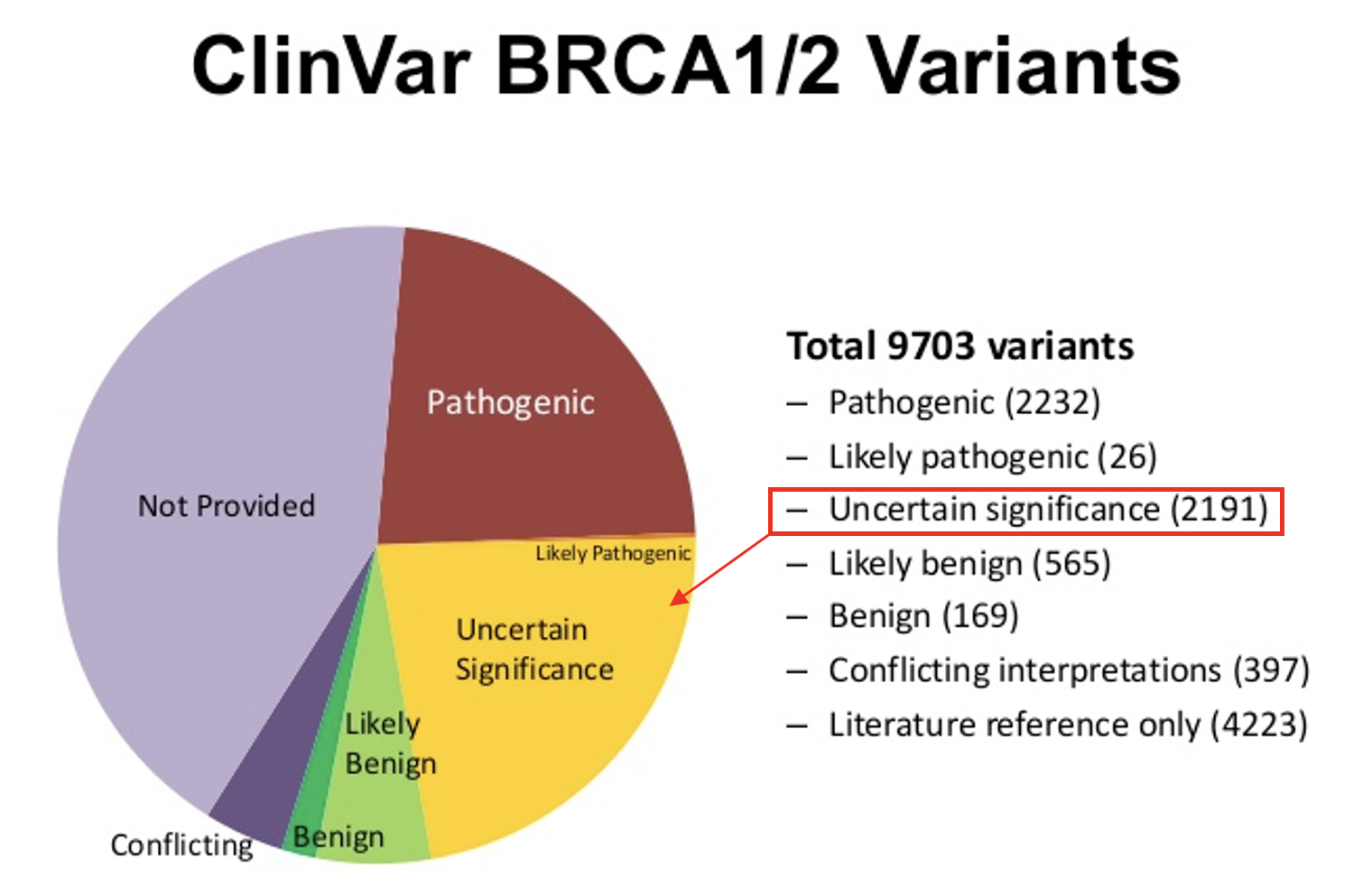
Esta problemática se origina atendiendo a diferentes causas, como la existencia de poca evidencia para clasificar la variante como causal o no, frecuentemente derivada por la ausencia de dicha variante genética en estudios poblacionales en los cuales se suelen reclutar pocos representantes de diferentes etnias, provocando errores en estimación de la frecuencia alélica en dichas cohortes.
Afortunadamente, la gran extensión de los estudios basados en secuenciación masiva de ADN está permitiendo que la cantidad de información obtenida de cada una de las variantes genéticas detectadas produzca un grado de exactitud mayor en la clasificación de variantes genéticas empleando las guías establecidas por ACMG.
Este sistema de clasificación tiene en cuenta diferentes características de las variantes genéticas, como su frecuencia poblacional, puntuaciones de patogenicidad estimadas por diferentes predictores genéticos (conservación, estabilidad proteica, etc), cosegregación de la enfermedad con la variante heredada de padres a hijos, etc. Todo ello hace que, cuanta más información disponible exista sobre la variante genética en estudio y se integre en este sistema de clasificación, obtengamos una resolución mayor en su clasificación patogénica, siendo precisa y certera, para el desarrollo de un diagnóstico clínico de cualquier enfermedad basado en estudios de secuenciación masiva.
Pero ¿cómo podemos mejorar la clasificación de las variantes genéticas? Actualmente, los investigadores que estudian la asociación de las variantes genéticas con enfermedades, nos recomiendan seguir una serie de pasos para contribuir a la disminución de esta problemática global, que en determinadas ocasiones llega a impedir un diagnóstico clínico certero y aumenta el retraso diagnóstico de manera considerable, llegando incluso a lograr cifras tan altas como ocurre frecuentemente en el angioedema hereditario, una enfermedad rara en la que se han publicado retrasos diagnósticos de más de 50 años en determinados pacientes. Para contribuir a la disminución de esta problemática mundial, los investigadores aconsejan:
Mantener actualizada la información publicada en la literatura relacionada con nuestra enfermedad, así como tener integrados los datos de diferentes repositorios utilizados globalmente.
Pueden tener nuevos datos que aún no hayan sido publicados y que pueden ser incorporados en los estudios de nuestra variante genética.
Actualmente, se han diseñado diferentes repositorios de información de acceso abierto, en las cuales los investigadores pueden registrar las variantes genéticas que estén estudiando y aportar los datos derivados de su investigación con el objetivo de otorgar la máxima información posible y obtener una clasificación precisa de la variante genética.
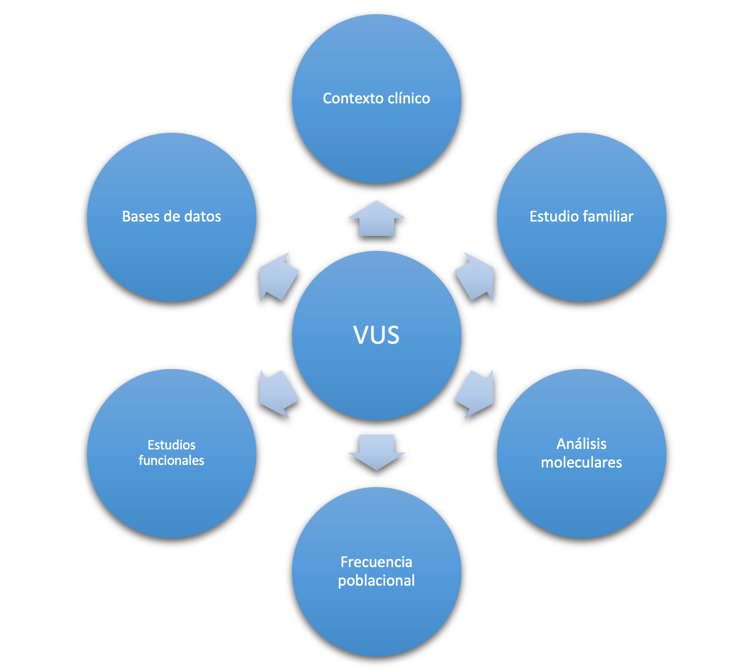
Como hemos visto, la integración de toda la información disponible sobre cada una de las variantes genéticas permitirá el agrupamiento de las mismas en las clases establecidas por ACMG, lo que a su vez facilitará la interpretación clínica y la asociación con la enfermedad será cada vez más exacta, todo ello con el objetivo de favorecer una rápida detección de la variante causal de la enfermedad y un diagnóstico más rápido y certero que permita prescribir un tratamiento eficaz, mejorando la calidad de vida del paciente.